Adopting a Technology-Driven Approach to Implementing EDC for Medical Devices
Benefits of business 4.0 approach in EDC in med-dev studies.
Clinical Investigation for safety and performance evaluation of Medical Devices differ from the Clinical Development of other medicinal products. These differences are related to data and tenure. Data is majorly collected in post-marketing phase, unlike drugs, that have clinical development carried out in three phases. The data in drug clinical trials is majorly related to patient health, while for devices, this data is about patient, the device the use of the device, and the device deployment procedure. While the large number of clinical research for drugs is mandatory, in medical devices it is only to support clinical evidence when clinical evaluation does not suffice to establish compliance with general safety and performance requirements; as evident in table 1.
Table 1: number of clinical studies logged for drugs and devices registered (as on Oct-2020 1-4)



Devices other than cosmetic and contraceptives cannot be tested in healthy human being. Hence, unlike drugs there are three types of investigations of the device depending upon the purpose of investigation i.e., pilot, pivotal and post-marketing. Pilot investigations are performed in a small population study to establish safety and performance of a product that is innovative or has critical risks. Commonly, this is the only clinical investigation performed for most devices before its marketing. A few devices require a Pivotal Clinical Investigation on a larger and diverse population, with end-point driven evidence generation. This investigation is performed for devices intended to treat critical diseases that has high risk associated with failure of the device. Major amount of data collection for such devices is generated in the Post Marketing environment. Regulatory Authorities are aware of this, and therefore there has been a significant surge in the number of Post Marketing Clinical Follow-up (PMCF) investigations after declaration of EUMDR-2017.
Medical device investigations are frequently compared with the drug development clinical phases( Phase I, II, and III). However, there is a wide difference between device and drug development. These differences percolate to all scientific aspects of clinical research including data collection, clinical data management (CDM), operations, statistics, and medical writing.
While the majority of CDM processes continue to be similar for all healthcare products, a few requirements are unique to the medical devices. Understanding these unique requirements for Case Report Form (CRF) designing and Electronic Data Capture (EDC) solutions is extremely important to successfully complete the clinical development process and market a safe and tested device.
There are two key challenges related to the design and implementation of a device EDC solution: 1: Regulatory requirements (Figure 1), and 2: Clinical aspects (Figure 2).
Complex regulatory requirements:
Medical devices must continually innovate. The products have to rapidly change and evolve to be more personalized and connected, leveraging AI and cognitive automation, robotics, and mobile technology. The constant changes and innovations on device developments, not only represent a big challenge for Life Science Industries and Manufactures, but also for Regulators.
New, evolving and complex regulatory requirements and government guidelines include changes pertaining to how manufacturers and distributors show the safety and efficacy of products (Medical Device Regulation, MDR, in Europe), demonstrate clinical evidence for all products as well as maintain systems and documents to remain compliant throughout the device lifecycle (Food and Drug Administration, FDA in the US). Both the U.S. FDA and Europe under MDR, demand more quality control processes throughout the device lifecycle including more audit trails.
Figure 1 regulatory requirements for CDM for medical devices

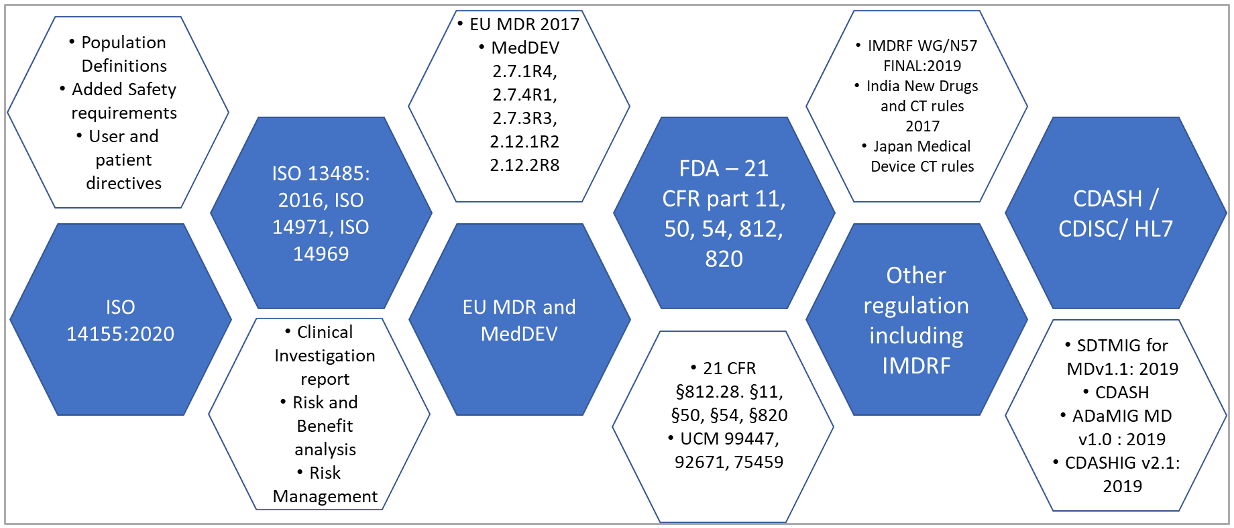

Device manufacturing organizations need to be responsive to regional and local Regulatory Authorities and government pre-requisites in an effective manner. Responding to the complex and dynamic regulatory environment requires the adoption of modern technology solutions for improved data capture, and safety process flexibility while maintaining compliance.
Complex clinical aspects:
The complexity of clinical data collected for Medical Device is another challenge for Device Manufacturers and for the clinical development of the product. This complexity is reflected mainly on the design of the CRFs. CRF related challenges include need for multi-level forms, multiple repeating tables, capability to handle data of multiple investigational and supporting devices and procedural data. Another complexity is in the device details, which may include one or more investigational devices with different sizes or variation, different combinations of supporting and accessory devices, etc. Often, accurate capture of operative and post-operative data affects the definitions of population like screened, enrolled, intent to treat and treated etc. In addition, some in-patient follow-up until discharge may alter the definitions of the outcomes. The challenge is more pronounced for procedure visit in interventionally deployed devices. Hence, the CRF designing based upon the disease, therapy, procedure, outcome criticality, and discharge related follow-up requires thorough understanding of the domain as well as clinical data management. This data is often populated by technical persons or specialized doctors, nurses and technicians. Hence, creating an interactive, intelligent query-based, high performance EDC system ensures that data is captured right at first time.
Figure 2 Data oriented challenges in medical device Clinical Data Management



Mitigating challenges in clinical data management with platforms and technology advances
Medical Device CDM challenges and subsequent EDC requirements are unique and distinct from the perspective of:
- device specific data: device identifiers, exposure, incidents, adverse events, tracking, device-subject interphase etc.
- procedure related data: procedure steps, time, complications, outcomes, effect, and patient-operator-procedure-device relationship etc.
- use of the device data: usability issues, instructions for use etc., and
- patient data: adverse events, adherence to handle and use of the device, demographics etc.
The collection of data from these factors influence the design of the EDC platform for effective implementation of Medical Device investigations. Some of these challenges and their process-technology-method trio solutions are provided in table 2.
Table 2 EDC Challenges and strategy for mitigation
Abbreviations; eCRF: electronic Case Report Form; ePRO: electronic Patient Report Outcome; AI: Artificial Intelligence, SDTM-MD – Structured data tabulation model for medical devices, ADaM-MD – Analysis Data Model for medical devices



Success of any study depends on an efficient, flexible and cost-effective platform, that has the capability to handle extremes of sample size, complex data structure, long duration and several sources of the data. This reinforces the need for a unified platform with optimum interoperability, scalability, flexibility and adaptive design, fully equipped to manage Medical Device trials with high operational efficiency (Figure 3). To implement an effective EDC solution for medical device and combination product studies, the actions can be planned in two states. In the current state, focus can majorly be on EDC management related process Improvement such as creation of standard CRF library for all reusable forms and tools, automation of CRF creation, Model Generation, Dashboards, ePROs etc.
The second action links to the fourth industrial revolution, also known as Business 4.0 accelerators, leveraging AI, Blockchain and Internet of Things (IoT/NFC), to create futuristic solutions enabling efficient CRFs, AI / IA automated tabulation and TFL generation, automation of Core Laboratory process and developing capability for Connected Data Collection through SMART contracts and Wearables.
Figure 3: Effective EDC: EDC functionality building blocks for medical device clincal data management NB: CCT and MIPOP are TCS brand names



Benefits of business 4.0 approach in EDC in med-dev studies
Reduced study startup timelines:
Automation of eCRF and edit checks with a data model helps reduce study start-up timelines leveraging Clinical Data Interchange Standards Consortium (CDISC-ODM) metadata driven approach. A well-defined and comprehensive library of eCRF’s and associated edit checks is amongst the strongest and standards-driven strategy to accelerate EDC development further.
Improved patient compliance:
Digitalization can help overcome one of the crucial KRI viz. Patient compliance in Medical Device trials. Adept user-friendly interface for patient engagement (Mobile apps, ePRO/Connected Clinical trials, SMART integration of devices) enable enhanced and sustained patient centricity (Visit reminders, SMS/email alerts).
Improved operational efficiency:
Use of cloud, block-chain and AI-based EDC systems integrated with Risk Based Monitoring (RBM) solutions can pave the way for improving overall operational efficiency. In addition, highly intuitive dashboards/graphics of key indicators and list of actions improve the user experience and performance by enabling real-time insights.
Data collection and submission:
The CDISC standards for Medical Devices have laid guidelines/standards, that are unique/specific to Device studies streamlining data submission to FDA. This too can be objectively managed if the EDC vendors can aim/target to make their tools/systems CDISC and FDA compliant.
Conclusion:
Implementing a Business 4.0 driven unified EDC platform for medical device trials can yield significant improvements in efficiency, standardization, data transfer accuracy thereby enabling sponsor to make more informed decisions. Effectively, the EDC system builds the sponsor’s roadmap to bring innovative and necessary medical devices to patients faster.
Dr. Ashish Indani is the Head of Research and Innovation, Dr. Alejandra Guerchicoff is the Industry Advisor, ADD Safety, Gaurav Srivastava is the Developer, Life Sciences Platform, Pratibha Potare is the Domain SME - Life Sciences Platforms, Sharad Sharma is the Domain Consultant- Life science & Healthcare, Clinical Data Management, Shivaji Bote is the Domain Consultant- Life science, Clinical Data Management, and Vandita Tripathi is the Head of ADD Data Management; all with TCS ADD Life Sciences platform at Tata Consultancy Services.
References:
- https://clinicaltrials.gov/ct2/search/advanced?cond=&term=&cntry=&state=&city=&dist=, searched on 20-Oct-20 at 9:55 AM IST
- https://www.isrctn.com/search?q=, searched on 20-Oct-20 at 10:12 AM IST.
- https://www.clinicaltrialsregister.eu/ctr-search/search?query=Drug, https://www.clinicaltrialsregister.eu/ctr-search/search?query=Device, searched on 20-Oct-20 at 10:21 AM IST.
- http://ctri.nic.in/Clinicaltrials/login.php Searched on 20-Oct-20 at 10:32 AM IST.
- International Standards Organization. (2020). ISO 14155 : 2020 Clinical investigation of medical devices for human subjects — Good clinical practice. International Standards Organization.
This article originally appeared in Applied Clinical Trials, January 2021.


 to FDA for Liberty endovascular robotic system")




